
For Physicians
Whole Transcriptome Sequencing
Whole transcriptome sequencing (WTS) uses the capabilities of high-throughput sequencing to gain insight into the RNA profiles of patient tumors. WTS provides extremely broad coverage and strong resolution (depth) into the dynamic nature of the transcriptome.
WTS enables gene fusion detection and RNA splice variant detection all from one streamlined test. The test identifies fusions independent of the breakpoints in DNA and has the ability to detect rare, or novel, fusion events better than targeted RNA sequencing or DNA-based methods. By providing broad exon coverage and capturing essentially all possible fusion partners, whole transcriptome sequencing can reliably detect different types of gene fusions, while reducing false positives caused by non-expressed rearrangements.
| Features | |
|---|---|
| Genes covered | Essentially All (23,000+) genes; clinically actionable genes will be highlighted on Caris Reports and unclassified RNA alterations will be available separately |
| Reads/Sample | 17 million reads |
| Assay method | Hybrid capture |
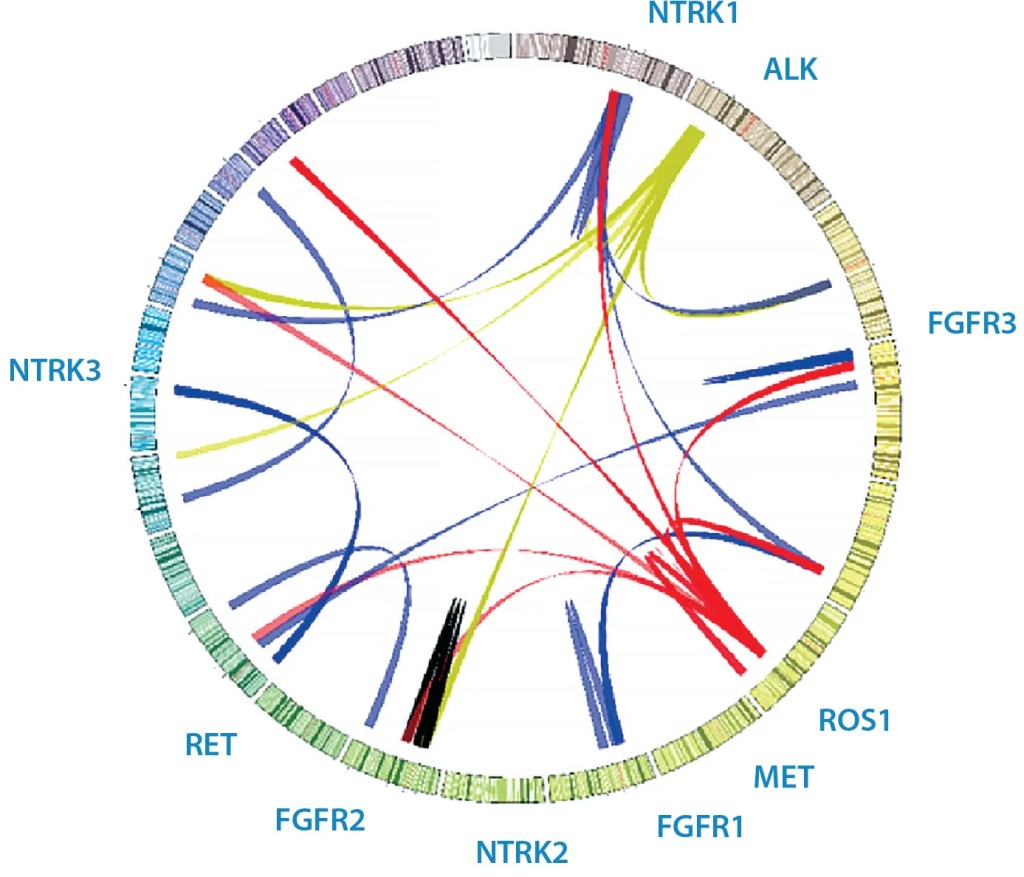
Figure 1: Sample Circos plot of various clinical fusions
Advantages of RNA Analysis via Whole Transcriptome Sequencing
RNA-based analysis, via Whole Transcriptome Sequencing, is the best method to detect clinically relevant aberrations, because the presence of RNA transcripts ensures that the alterations have been transcribed from DNA to RNA. MI Transcriptome also has the potential to discover previously uncharacterized events, which is important when identifying patients who may derive a strong response to targeted therapy. The test uses the same tissue requirements as Caris’ DNA tumor profiling and delivers results in the same turnaround time.
Variant Transcripts
- Caris RNA sequencing picks up variant transcripts by looking at the spliced mRNA directly
- RNA sequencing provides more relevant information when analyzing biological functions; Caris testing will provide an estimate of gene expression levels to feed pathway analysis tools
| Fusion Analysis Method | # of Genes | Clinical Actionability | Fusion Expression |
|---|---|---|---|
| RNA Sequencing | Essentially all genes | Can detect gene fusions | Can identify if fusion is expressed |
| DNA Sequencing | Limited (hot spots) | Can detect rearrangements that may not always result in gene fusions | Cannot identify if fusion is expressed |
Figure 2: Diagram showing advantages of RNA Sequencing over DNA Sequencing for fusion analysis
Example: An ALK fusion may be found at the DNA level but it may not be expressed as RNA. Some fusion partners such as EML4 result in dramatic overexpression of ALK and increase in its kinase signaling. However, other fusion partners may not result in ALK overexpression and thus would not potentiate the oncogenic ALK kinase activity. Such cases are unlikely to derive significant benefit from targeted ALK therapy.
RNA is the Superior Method of Gene Fusion Analysis
Caris molecular profiling includes MI Transcriptome™, Whole Transcriptome Sequencing (WTS), via RNA next-generation sequencing. MI Transcriptome enables gene fusion and splice variant detection from one streamlined test. RNA testing identifies fusions independent of the breakpoints in DNA and, therefore, has the ability to detect rare, novel fusion events better than DNA-based methods.
DNA vs RNA – Direct Comparison Shows RNA is the Superior Method for Fusion Analysis
A recent study by Benayed, et al.1 shows that RNA fusion analysis2 identifies additional alterations as compared to DNA fusion analysis3.
Highlights include:
- 2,522 NSCLC cases were sequenced using standard DNA-based methods. Of those, 232 cases lacked an oncogenic driver.
- These 232 driver-negative cases were then RNA sequenced and 36 (15.5%) were found to have oncogenic drivers that were not detected by DNA sequencing, 33 of which were clinically actionable.
- Importantly, in this analysis DNA sequencing did not detect any fusions in NTRK 2/3 that RNA was able to detect.
- The data from Benayed, et al. are significant, but still limited by the use of targeted RNA sequencing. Further investigation using WTS would likely identify even more fusion partners.
| NSCLC NCCN guidelines suggest using RNAseq to maximize the number of fusions detected5 | |
|---|---|
| Fusion/Variant | # detected by RNA seq not detected by DNA seq |
| ALK | 4 |
| BRAF | 1 |
| FGFR2 | 1 |
| MET (exon14) | 6 |
| NRG1 | 5 |
| NTRK2 | 1 |
| NTRK3 | 2 |
| RET | 3 |
| ROS1 | 10 |
N = 232
RNA-Based Fusion Analysis of NTRK1/2/3 Finds 50% More Clinically Actionable NTRK3 Fusion Partners than DNA
In a review of the Caris database of over 30,000 cases run for RNA fusion detection, additional fusion partners were found beyond what other commercially available DNA panels report. In this review, a DNA only approach, such as FoundationOne®CDx, would have missed 50% of clinically actionable NTRK3 mutations, due to the use of ETV6 (only), a common fusion partner for NTRK3*. Using RNA based fusion detection, multiple other clinically actionable NTRK3 fusion partners were found4.
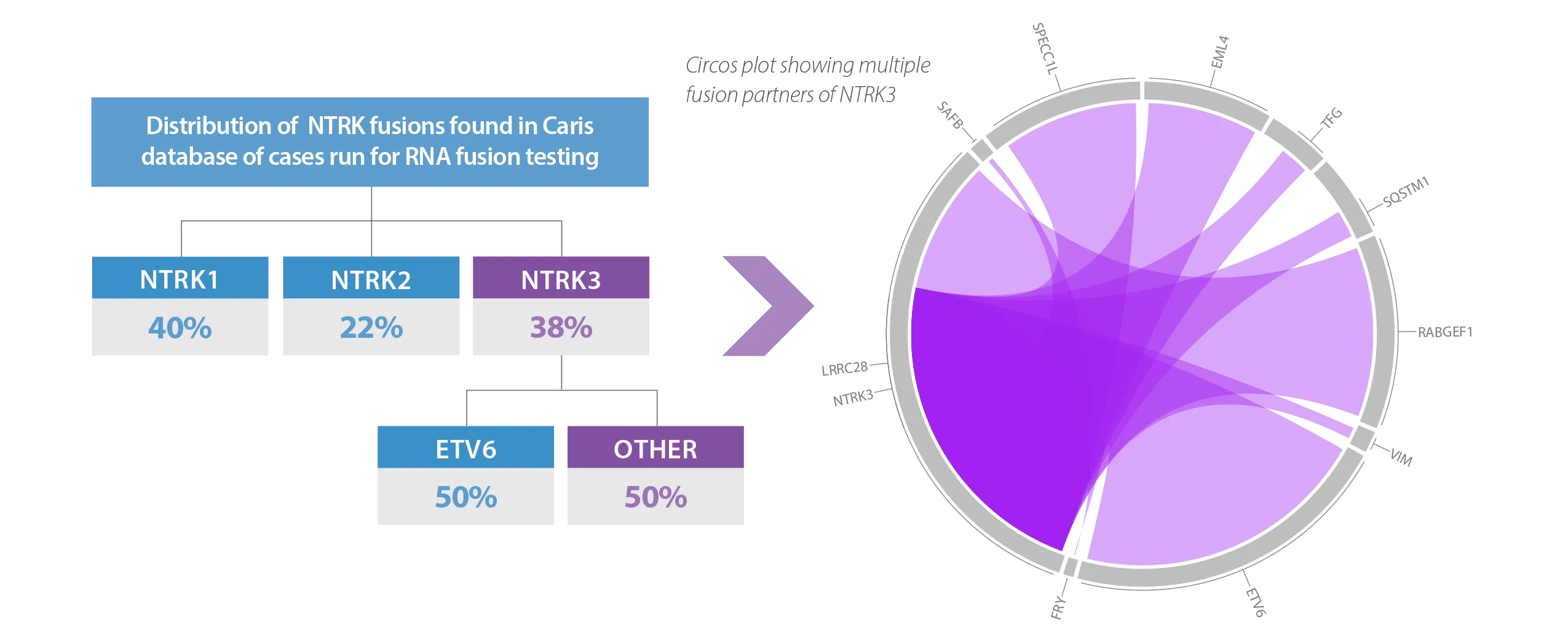
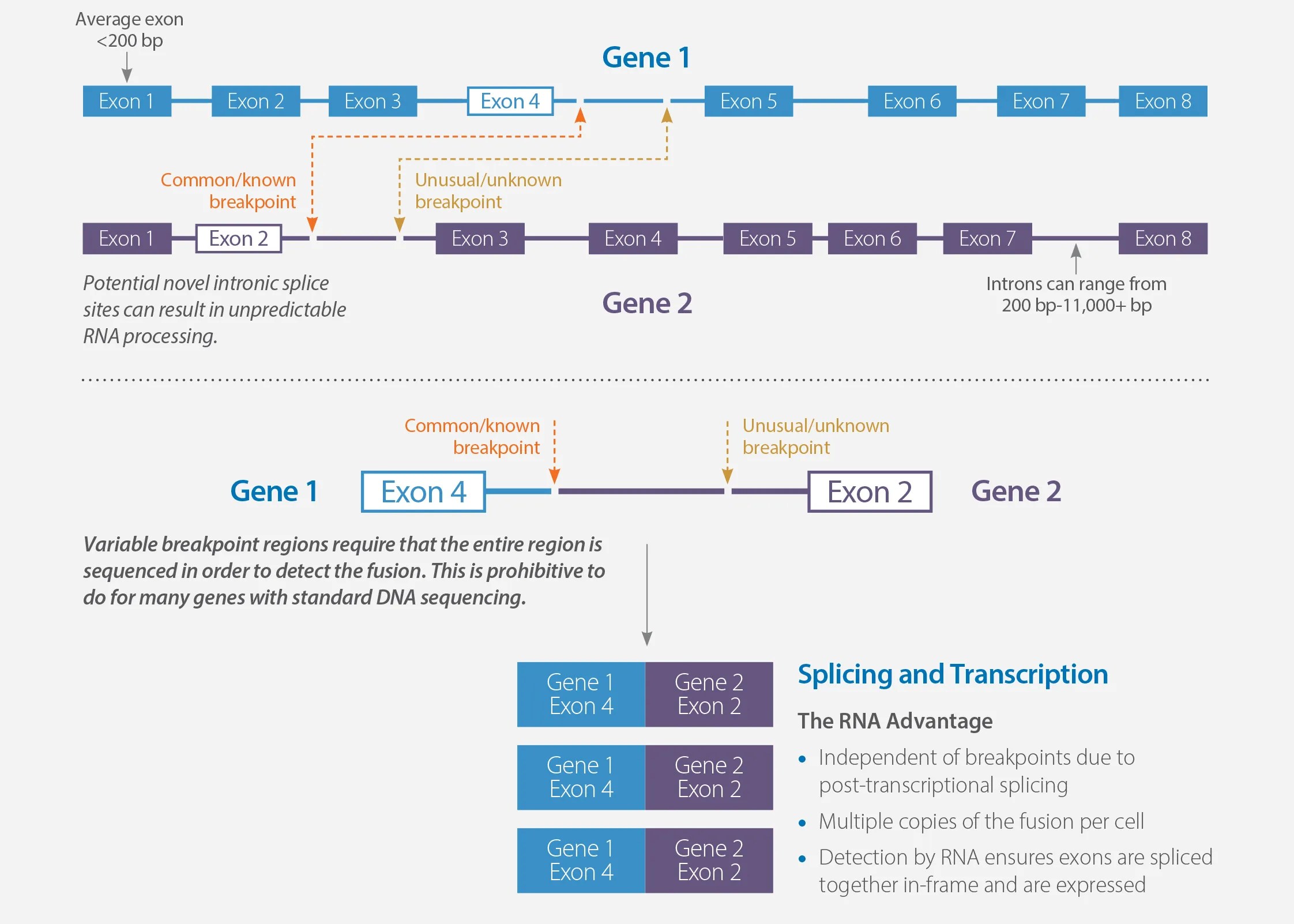
Whole Transcriptome Sequencing Captures All Potential Fusion Partners
MI Transcriptome can reliably detect expressed missed gene fusions (false negatives) and reduce false positives caused by unexpressed rearrangements called out using DNA-based fusion detection. Gene fusions are formed when exons from one gene rearrange to connect with exons from another gene. The breakpoints for these rearrangements frequently occur in the introns between the exons of the genes.
*FoundationOneCDx (Source: https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019C.pdf ) 1. Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no driver alteration detected by DNA sequencing and low tumor mutation burden. Clinical Cancer Research, 2019. 2. MSK Fusion panel (Targeted RNA) 3. MSK-IMPACT™ (DNA) 4. Internal Data, Caris Life Sciences Database 5. National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer Version 1.2020. 2019 Nov 6;National Comprehensive Cancer Network.
Complete Gene Coverage with Caris Molecular Testing
As the pioneer in precision medicine, Caris was the first to provide comprehensive Whole Exome and Whole Transcriptome sequencing for every patient. Each Caris molecular profiling order includes next-generation sequencing of all 23,000 genes.
Looking for a specific gene? Use the full gene search below to verify that it is included in Caris profiling, or browse the list of genes most commonly associated with cancer.
| Whole Transcriptome Sequencing – Genes most commonly associated with cancer listed below. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Fusions (RNA) | Variant Transcripts (RNA) | |||||||
| ABL | BRD3 | FGFR3 | INSR | MYB | NUMBL | PRKCA | RSP03 | AR-V7 |
| AKT3 | BRD4 | ERG | MAML2 | NOTCH1 | NUTM1 | PRKCB | TERT | |
| ALK | EGFR | ESR1 | MAST1 | NOTCH2 | PDGFRA | RAF1 | TFE3 | EGFR vIII |
| ARHGAP26 | EWSR1 | ETV1 | MAST2 | NRG1 | PDGFRB | RELA | TFEB | |
| AXL | FGR | ETV4 | MET | NTRK1 | PIK3CA | RET | THADA | MET Exon 14 Skipping |
| BCR | FGFR1 | ETV5 | MSMB | NTRK2 | PKN1 | ROS1 | TMPRSS2 | |
| BRAF | FGFR2 | ETV6 | MUSK | NTRK3 | PPARG | RSPO2 | ||
Discover
More
Caris tissue-based profiling utilizes the most advanced technologies and comprehensive approach to analyze DNA, RNA and proteins to reveal a molecular blueprint that guides more precise and individualized treatment decisions that leads to improved patient outcomes.
Caris Assure is a minimally invasive blood test that provides comprehensive molecular analysis of tumor biomarkers when tumor tissue is not feasible.